

差异基因表法分析
DESeq2所需数据R代码输出数据备注原始Counts数据样本分组信息123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960616263646566676869707172737475767778798081828384858687888990919293949596979899100101102103104105106107108109110111112113114115116117118119120121122123124125126127128129130131132133134135136137138139140141142143144145146147148149150151152153154155156157158159160161162163164165166167168169170171172173174175176177178179180181182183184185186187188189190191 ...

Mac终端:服务器与本地文件交互
从服务器下载文件到本地
打开Mac终端
使用scp命令下载文件
命令💻参数解释🗣️1scp -r xiaosu@10.10.140.xxx:~xiaosu/gorden /Users/xiaosu/Desktop
参数
说明
-r
如下载的是文件夹需使用此参数,如果是文件可不使用此参数
xiaosu@10.10.140.xxx
服务器地址
~xiaosu/gorden
输入服务器中要下载的文件路径
/Users/xiaosu/Desktop
输入本地地址,必须是全局路径

生信数据格式
GENBANK简介常用代码GenBank是一个由美国国家生物技术信息中心(NCBI)维护的数据库,它是一个免费的数据库,包含了大量的核酸序列和蛋白质序列。这些数据包括了基因组、mRNA、EST、蛋白质等。GenBank数据库中的数据是以文本的形式存储的,每一条序列数据都有一个唯一的标识符,这个标识符是一个以“LOCUS”开头的行,后面跟着这条序列的名字。GenBank数据库中的数据是以一种叫做GenBank格式的格式存储的,这种格式是一种文本格式,它包含了序列的名字、序列的长度、序列的来源、序列的特征等信息。
例如文件NC_045512包含以下信息:1234567891011121314151617181920LOCUS NC_045512 29903 bp ss-RNA linear VRL 18-JUL-2020DEFINITION Severe acute respiratory syndrome coronavirus 2 isolate Wuhan-Hu-1, complete genome.ACCE ...
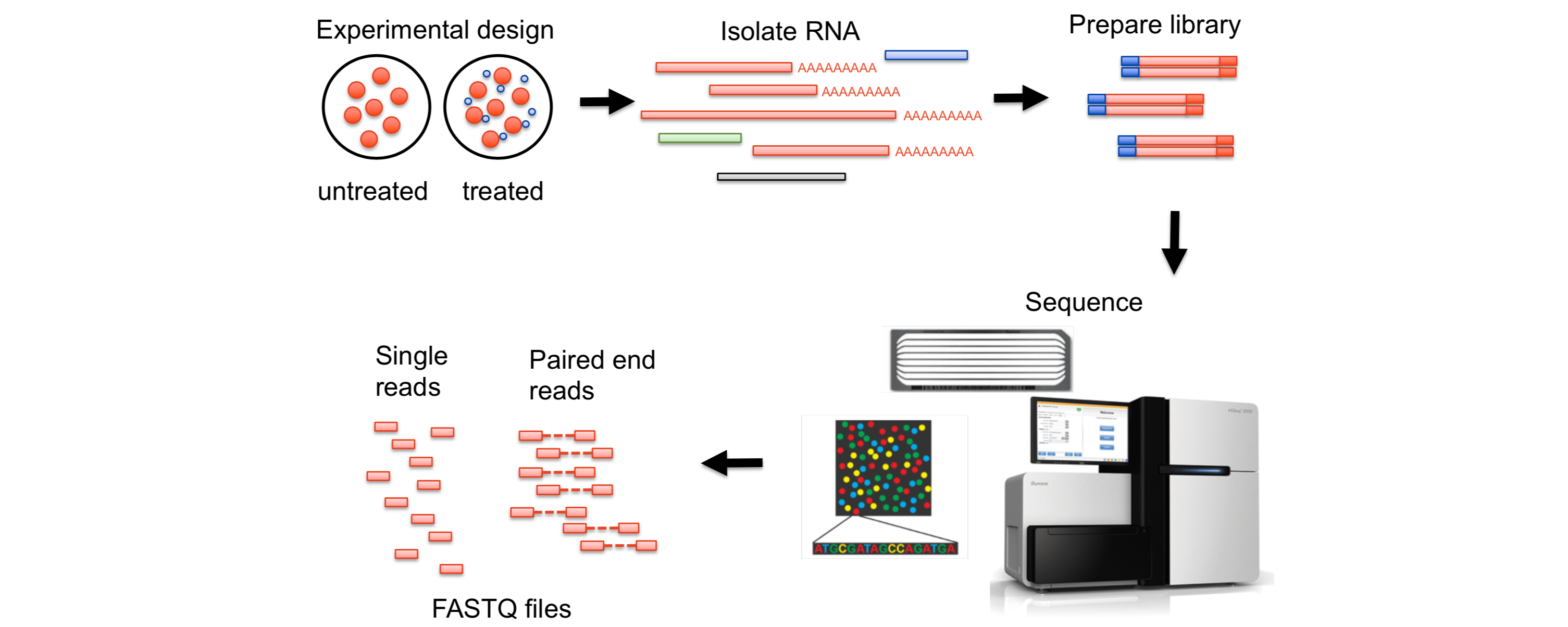
RNA-Seq
RNA-Seq指南RNA-Seq参考根据基因组进行量化RNA-Seq根据基因组进行量化时,参考数据是基因组序列,基因组注释文件是基因组的注释信息,包括基因的位置、外显子的位置、基因的功能等信息。该方法将对齐文件与注释相交叉,以产生“计数”,然后过滤这些计数以保留统计上显著的结果。定量矩阵代表对齐与标记为转录本一部分的区间重叠(交集)的次数。
在通过参考基因组和注释信息对读段进行映射,然后再进行表达量估算时候,又有两种不同的基因表达量分析方法。
基因水平分析:
这种分析方法把一个基因的所有转录本(即由基因的不同区域(外显子)组成的不同版本)视为一个整体。
在这种分析中,不区分各个不同的转录本,而是将它们合并为一个单一的序列,这个序列包括了该基因的所有外显子。
这个合并的序列被认为是“代表性”的,但它可能并不对应于任何实际存在的单一转录本,它更像是一个虚拟的总和。
对于某些研究目的而言,这种简化的方法足够用来获取有意义的数据,尤其是当研究的重点是基因整体活动水平时。
转录本水平分析:
与基因水平分析不同,转录本水平分析关注于每个单独的转录本。
这种方法不会将不同的转录本合并 ...
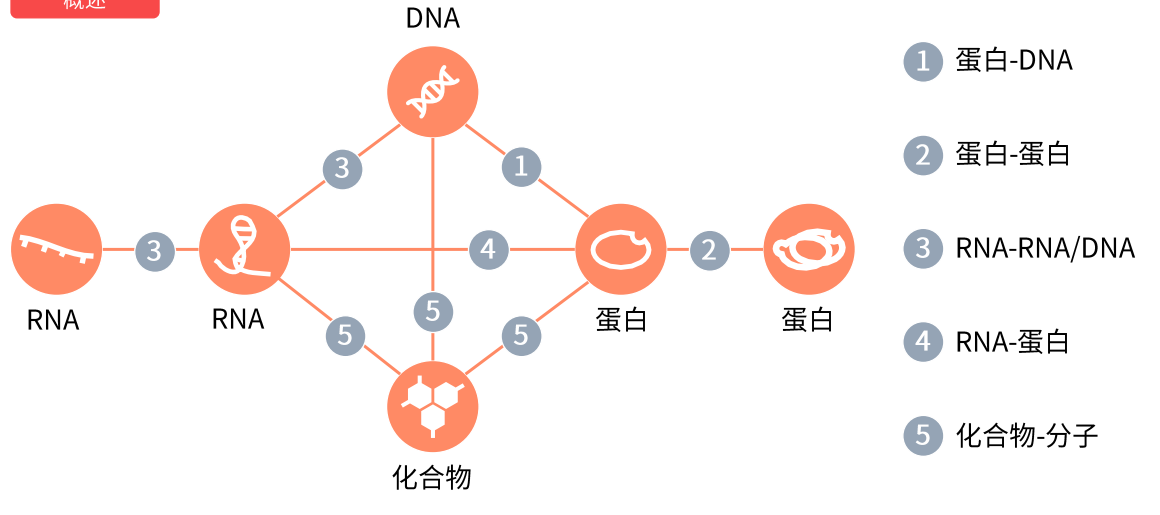
分子互作
蛋白-DNA
也称转录因子模式,所有转录因子都包含一段 DNA结合域,结构域能够识别和结合基因启动子特征DNA序列(motif) ,进而影响靶基因从 DNA 转录产生 RNA 的过程,效应结果是 RNA量的多少变化,如果这个RNA是可以编码蛋白的mRNA,那么最终就会造成靶基因蛋白量的上调或者下调。
转录因子上的氨基酸序列和结构结合的 DNA 序列是有一定规律的,这种规律是预测转录因子调控靶基因的理论基础。
启动子是一段DNA序列, 启动子里含有 RNA 聚合酶的特异性结合序列,同时可以跟转录因子结合控制基因的转录活性。
相关数据库
转录因子转录因子预测原理研究思路预测数据库转录因子验证实验纯生信:
根据测序结果筛选出的关键基因,探索能否找到潜在的转录因子调控这些基因
可以同时引入与转录因子和靶基因结合的非编码RNA,构建调节网络
或者识别出的差异表达基存在转录因子,可以以这个转录因子为中心,构建调节网络
基于转录因子本身的的表达水平、突变状态、甲基化水平、做转录因子与表型的生信分析
生信+实验:
基于序列保守性对互作关系进行预筛选,然后再通过EMSA、ChIP等实验验证 ...
WGCNA
WGCNA 入门背景知识简介原理基本分析流程计算步骤
权重(weghted):基因之间不仅仅是相关与否,还记录着它们的相关性数值,数值就是基因之间的联系的权重(相关性)。
模块(module):表达模式相似的基因分为一类,这样的一类基因成为模块。
Eigengene:基因和样本构成的矩阵。
邻接矩阵(Adjacency Matrix):是图的一种存储形式,用一个一维数组存放图中所有顶点数据;用一个二维数组存放顶点间关系(边或弧)的数据,这个二维数组称为邻接矩阵;在WGCNA分析里面指的是基因与基因之间的相关性系数矩阵。 如果用了阈值来判断基因相关与否,那么这个邻近矩阵就是0/1矩阵,只记录基因相关与否。但是WGCNA没有用阈值来卡基因的相关性,而是记录了所有基因之间的相关性。
拓扑重叠矩阵(TOM,Topological Overlap Matrix):WGNA认为基因之间的简单的相关性不足以计算共表达,所以它利用邻近矩阵,又计算了一个新的邻近矩阵。一般来说,TOM就是WGCNA分析的最终结果,后续的只是对TOM的下游注释。
WGCNA(Weighted Gene Co-expres ...

生物数据库基因ID
小苏碎碎念:
科研分析中经常会遇到各种各样的基因ID类型,了解各种命名的来源和规则有助于我们在不同的数据库之间进行转换和使用。同时因各种各样的数据库越来越多,本文档也会持续完善和更新~~~🍊。
背景知识
NCBI:National Center for Biotechnology Information, 即美国国家生物技术信息中心,是一个机构名称,不是数据库。
Entrez 是一个归属于 NCBI 的综合性的生物信息数据检索引擎系统,这个搜索引擎整合了包含核酸、蛋白质、基因、基因组、GEO 等在内的很多常用数据库。
HGNC: HUGO Gene Nomenclature Committee, 即人类基因命名委员会。人类大多基因的命名,是有HGNC完成的。
小鼠(mouse)的基因命名来源于MGNC;大鼠(rat)基因命名来源于RGNC;斑马鱼(zebrafish)基因命名来源于ZFIN
Ensembl 基因组数据库项目,是 1999 年启动的,来应对当时即将完成的人类基因组计划的一个科学项目,是科研人员用于检索基因组信息的最常用数据库之一。
refseq参考序列数据库,是NC ...

定量引物设计
查找基因进入NCBI官网
查找序列
查找基因ID
引物设计打开Prime-BLAST
根据需求填写参数
结果筛选

tidyverse:处理关系数据
文档使用数据源说明
文档中举例除了使用自建数据外,我们需要使用 nycflights13。这个数据包含了 2013 年从纽约市出发的所有 336 776次航班的信息。该数据来自于美国交通统计局, 可以使用?nycflights13 查看其说明文档。包括flights, airports, planes, weather, airline五个数据框。 文档中如出现此类数据对象,不再另行说明。flights: 包含航班信息airlines:可以根据航空公司的缩写码查到公司全名。airports:给出了每个机场的信息,通过 faa 机场编码进行标识。planes:给出了每架飞机的信息,通过 tailnum 进行标识。weather:给出了纽约机场每小时的天气状况。R包使用:12library(tidyverse)library(nycflights13)
相关概念
键:用于连接每对数据表对变量称为键,键是能唯一标识观测的变量(或变量集合)
主键:唯一标识其所在数据框的观测
外 ...

GEO数据库
GEO数据库 写在前面的话
GEO 数据库是 NCBI 网站下的子数据库,主要收录基于芯片和测序技术的数据,来源于全球众多研究者的上传数据。GEO数据库一直是进行生信挖掘的重要数据库,了解GEO数据库的数据形式,组织类型及下载方式进行数据挖掘的必备技能。
数据存储格式SOFT 格式SOFT (Simple Omnibus in Text Format)格式,是一种紧凑、简单、基于行的 ASCII(美国信息交换标准代码)文本格式, 包含实验数据和注释数据。
MINiML 格式MINiML (MIAME Notation in Markup Language, pronounced minimal)格式,与 SOFT 格式文件的包含信息完全相同,仅格式不同,为 XML 格式。 XML(eXtensible Markup Language)又称为可扩展标记语言,极其简单,使其易于在任何应用程序中读写数据。MIAME 和 MINSEQE 分别是芯片和测序数据上传的标准指南,指出上传的芯片或测序数据最起码应包含的数据内容。
Series Matrix filesSeries Matrix fil ...





